Research
The main goal of our research group is to understand how light can be used to control chemistry. To that end, we develop theoretical methods based on quantum mechanics that can be used to simulate light-matter interactions within a computer. Some of our current interests are:
1. Modeling charge migration with high-level electronic structure methods
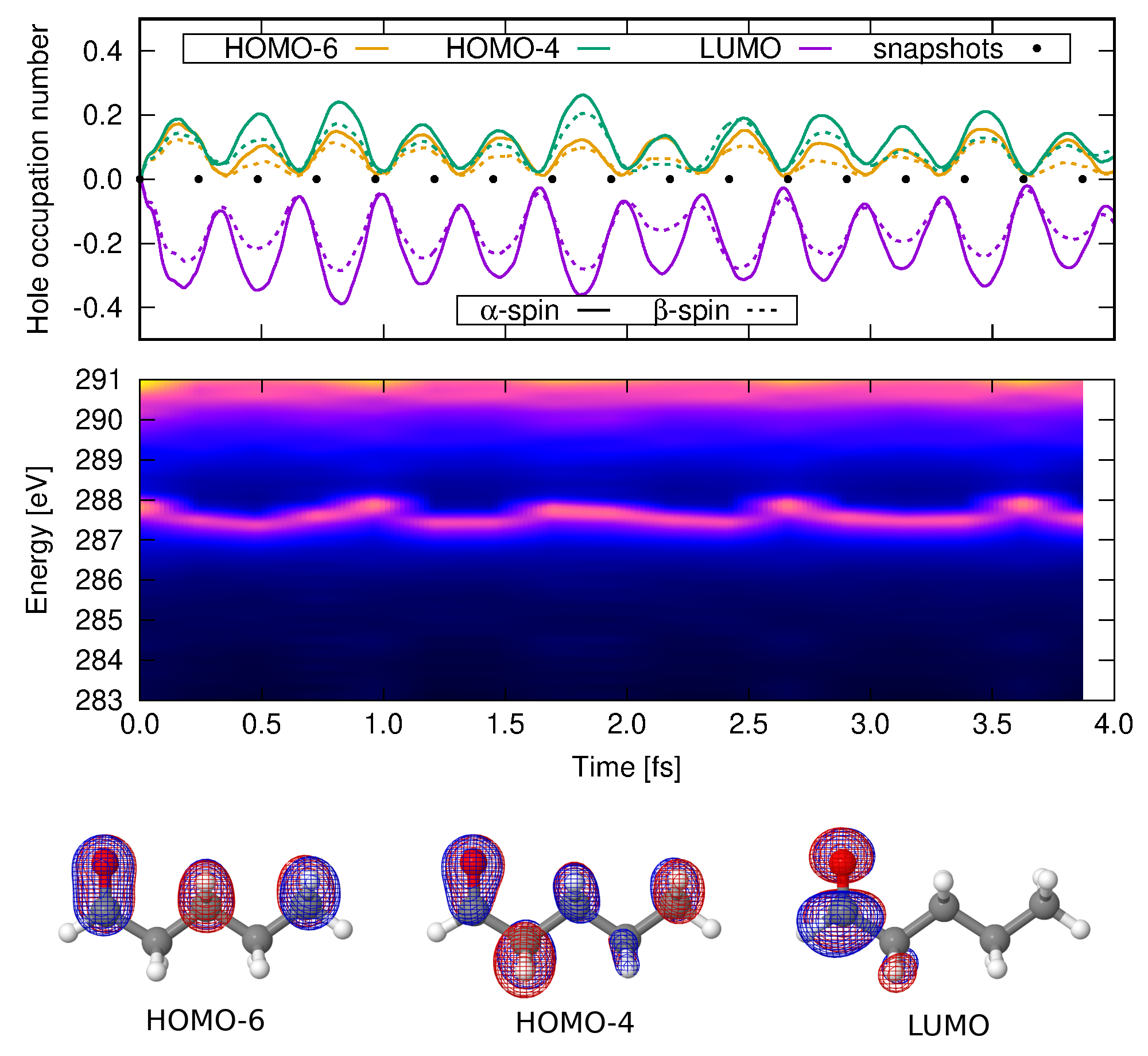
When a molecule is photo-ionized, the newly created hole quickly initiates a fast charge migration through the molecular backbone. This migration happens in femto- to sub-femtosecond time scales, preceding any structural reorganization and is driven mainly by electron correlation. The possible pathways in which hole migration takes place leads to eventual hole-density accumulation at specific sites in the molecule, which coupled with nuclear motion, determines photochemical outcomes. Thus, being able to model ultrafast charge migration with high-level electronic structure methods with a reasonable computational cost is an important step for a better understanding and control of phtochemistry.
2. Exploring novel spectroscopies in the X-ray regime
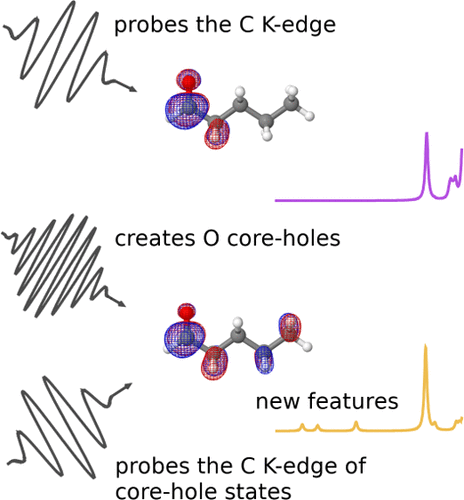
The interaction of molecules with high-intensity X-rays can lead to the formation of core-hole states, due to the ionization of core-electrons. These core-hole states are highly sensitive to their molecular environment, and thus, can be used as a probe for chemical change. Exploring how the formation of core-hole states affects the electronic structure of interesting molecular systems offers an invaluable opportunity to discover new spectroscopic signatures and spectroscopies.
3. Modeling the interaction of molecules with electric fields within confined environments
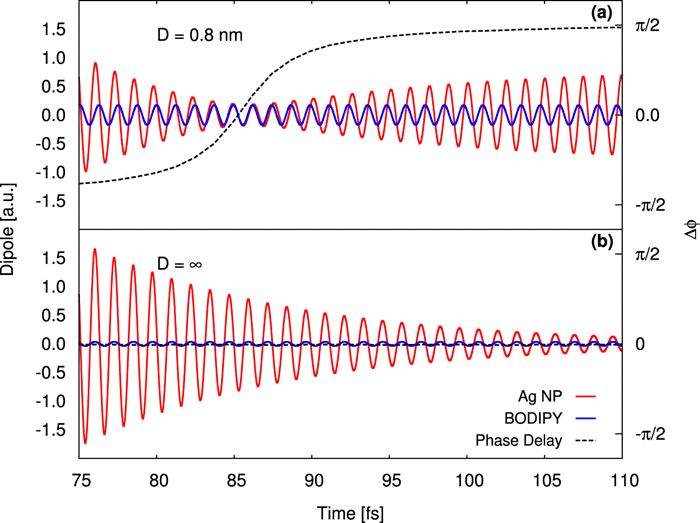
The interaction of molecules with electric fields in confined environments, such as in localized plasmons and optical cavities, can lead to a drastic change in the molecular electronic structure. Such changes can be large enough to promote new chemical reactions. An important step towards a better understanding of how these changes can be used to mediate new chemistry is the ability to perform computational simulations that go beyond the semi-classical treatment of electric fields in a reliable and inexpensive way.
4. Development of efficient algorithms for the description of spectroscopic properties with high-level electronic struture theories

The accurate description of spectroscopic properties is of absolute importance in quantum chemistry and can only be consistently achieved through high-level electronic structure methods. Nonetheless, the elevated computational cost associated with these methods limits their applications to rather small molecules. Hence, the development of efficient algorithms that can facilitate the simulation of large molecules is a major challenge and a concerted effort in quantum chemistry.
5. Simplified methods for modeling photo-damage in biologically and environmentally relevant molecules
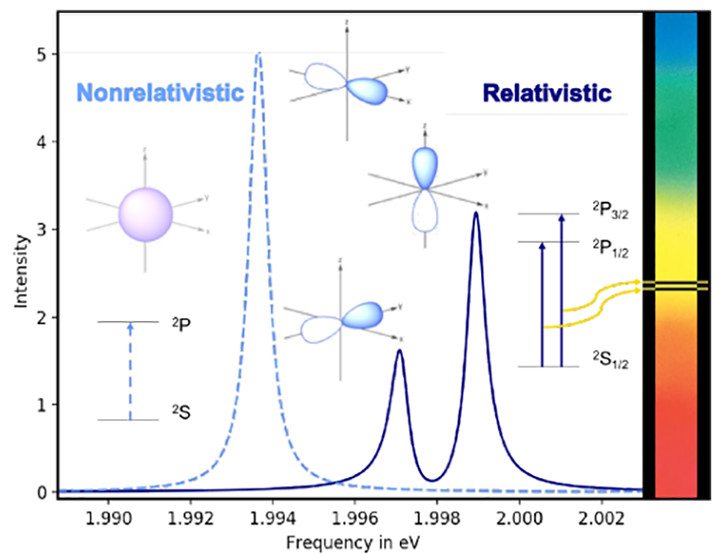
6. Simplified approaches for introducing relativistic corrections within time-dependent electronic structure theories